1. 受体学说
1.1 受体学说的提出
现代药理学认为,药物是通过结合体内靶标从而产生药理效应的。广义的靶标包括受体、酶、转运体、离子通道、遗传物质等。靶标的概念源自于受体学说。
德国医生Ehrlich首先提出“化学受体”这一概念,将药物与受体的作用描述为“化合物嵌入受体复合物中”,并指出“如果一个化合物能高度选择性地靶向疾病组织,即能对疾病组织产生毒性,专门杀死疾病组织。
英国科学家Langley在研究毛果芸香碱和阿托品对唾液分泌的拮抗作用时,提出了“受体”概念。他为神经末梢细胞上存在某种物质,能与这2种化合物形成复合物;这种物质是机体接收和产生信号的开关,可以被某些化合物激活或关闭。
英国药理学家Clark是现代受体理论的创始人,同时也是定量药理学的创始人。他指出:“如果不假设药物与受体以一种特殊的方式结合在一起,就不能解释我们所看到的非凡的药物效应。“
1.2 药物-受体相互作用的定量描述
1.2.1 Hill方程
英国科学家Hill在研究氧气和血红蛋白结合饱和度与氧气压力的平衡关系时,提出了一系列的方程,用以描述氧气-血红蛋白结合率()、氧分压()、氧气-血红蛋白结合常数()之间的关系,其中一个方程的形式如下:
1.2.2 Law of mass action
上述的Hill方程只是一个拟合实验结果得到的经验公式。Clark等利用质量作用定律讲改公式进行了推导。
质量作用定律的定义是:化学反应速率与反应物的有效质量成正比,其中的有效质量实际是指浓度。它可以确定化学反应中各反应物和生成物的活性质量之间的联系,在化学平衡学说中具有重要的意义。
1.2.3 Hill方程的推导
设受体为,体内的受体总数为,向体内注射药物分子;药物会与受体结合成复合物,复合物也会解离成游离的药物和受体;时刻的药物浓度、受体浓度、药物-受体复合物浓度分别为、、;结合与解离的可逆反应的结合速率常数为,解离速率常数为,平衡常数。
当体内受体全部被药物结合时,产生的药理效应最大,为100%。显然药物只能与受体部分结合,则此时效应与药物-受体结合率(即受体占有率)成正比。
根据质量作用后定律,写出药物与受体的结合达到平衡时,药物-受体复合物的浓度随时间的变化:
解以上方程,得:
可以发现上式即为时的Hill方程。如果一个药物分子最多只能与一个受体的结合,可以使用该表达式。
事实上,很多时候药物与受体结合并不是,而是,其中称为Hill系数,它会改变曲线的形状,此时的Hill方程为:
一般情况下,我们都会使用带有Hill系数的Hill方程对药物的量-效关系进行描述。
值得一提的是,生物化学描述酶促反应动力学的Michaelis-Menten方程,也是Hill方程的特例;人口学中描述人口增长的logistic分布,也与Hill方程的曲线相似。究其本质,都是由于某一过程存在“饱和”现象,导致曲线不会增长到无穷大,而是趋近于某一条渐近线。
1.3 其他模型
1.3.1 竞争抑制模型
在Hill方程的基础上,Clark等提出竞争抑制模型,表示2个配体与同一受体的竞争性结合。1947年,英国药理学家Schild将该模型用于实际,发展出了Schild回归和Schild图,提出了表征药物强度的方法。
1.3.2 Ariens的模型
很据Hill方程,药物的效应等于其对受体的占有率(receptor occupancy,RO),但是经常发现不同的药物,作用于相同的组织,产生的药理效应是不同的。这个现象导致了受体的“完全”和“部分”激动的概念。
1954年,德国药理学家Ariens引入了“intrinsic activity”这一参数,用以解释不同药物作用于同一受体而产生不同效应的现象。他认为这个参数取决于药物本身。
1.3.3 Stephenson的模型
很多时候,药物的效应并不是与受体占有率成正比的,这表明除了药物结合受体之外,可能还存在其他的影响药效的因素。
1956年,英国药理学家Stephenson在一篇论文中提出,药理效应与机体受到的刺激成正比,由此引入了“efficacy”这个参数。他认为药理效应是药物的受体占有率与efficacy的乘积,efficacy的值取决于不同的组织本身,而与药物无关。
模型的公式如下:
其中,为药物与受体的结合常数,为药物浓度,为药物刺激受体产生效应的诱导系数,即以上提到的“efficacy”这一参数。该公式与Hill方程相比,仅增加了诱导系数。
1.3.4 Furchgott的模型
1966年,Furchgott提出“initial concentration of total active receptors”这个隐含参数,并纳入到Stephenson的模型中,他认为Stephenson所提出的“efficacy”也归结为这个参数。此外,他还提出了“intrinsic efficacy”这一参数,用来定义完全取决于药物本身的那部分药效。
至此,经典的受体理论将药物的药理作用分为2部分:(1)取决于药物的部分,可以用药物结合受体的亲和性及内在活性来定义;(2)取决于机体的部分,可以用受体总浓度及刺激作用来定义。
1.4 传统受体学说的局限性
Hill方程与Stephenson方程只适用于药物的药效(response)与药物和受体的结合成正比的情况,即有结合就产生的药效。但是很多时候,药效与药物-受体相互作用的复杂过程密切相关,如受体构象的变化、药物对受体构象的选择性、受体下游分子的调控等等。
所以到目前为止,依然没有一个公式可以将药物与受体的相互作用及产生药效的机制完全参数化,直到20世纪80年代分析药理学的产生。
2. 分析药理学
分析药理学由英国药理学家Black建立,它可溯源于Black和Leff在1983年合作发表的一篇论文,该文提出一种新的药理学研究策略——操作模型(operational model)。分析药理学可以定义为“使用操作模型研究药物作用机制的学科”。
2.1 分析药理学的研究流程
分析药理学使用假设性机制来解释药物作用的机制,主要包括如下步骤:
(1)通过实验得到量-效曲线;
(2)提出多种药物作用机制;
(3)根据假设的作用机制,得到理论量-效曲线;
(4)比较实验量-效曲线与理论量-效曲线,如果相符,则该机制即为可能的药物作用机制;
(5)如果实验结果与理论结果不符,则需要分析原因,提出新的机制,直到获得与实验相符的机制。
2.2 操作模型
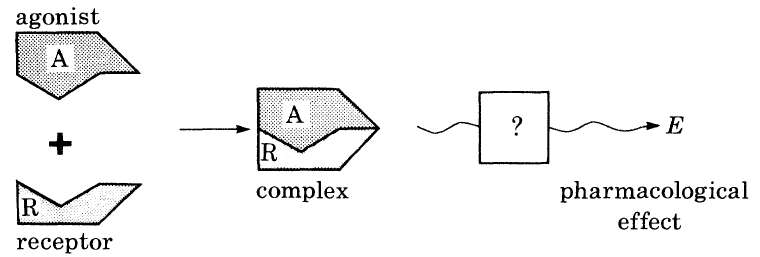
Black1983年发表了关于激动剂的操作模型。如下图所示,药理效应的产生分为2部分:激动剂(A)与与受体(R)结合成复合物(AR);复合物产生效应(E)。
2.3 E-A的直角双曲线关系
2.3.1 E-A关系
很多E-A曲线都显示效应随着激动剂的浓度的增加而增加,至少需要3个参数来进行表征:激动剂饱和时产生的最大效应、达到最大效应的一半所需激动剂浓度、E-A曲线在一半处的斜率。我们将常见的E-A曲线称为“直角双曲线”,其表达式如下:
至于为什么称为直角双曲线,因为E与A的关系实际上是由一个反比例函数经过平移得到的,而反比例函数的图像一定是双曲线。我们考虑如下反比例函数:
如果我们将该函数的图像向上平移个单位,向左平移个单位,则函数表达式为:
经整理得:
若,则
就得到了E-A曲线的表达式。因此E-A曲线是由反比例函数的图像经平移后得到的。
2.3.2 AR-A的关系
如上图所示,激动剂与受体的结合达到平衡状态时,满足以下关系:
化简得:
分子与分母同时除以,令,得:
其中,这个参数表征了药物对受体的亲和力。可以发现,AR与A的关系也是一个直角双曲线。
2.3.3 E-AR的关系
我们还需要建立从AR到效应的关系:
事实上,从AR到E的过程是一个黑盒子。我们知道,AR-A关系是一条直角双曲线,如果E-A关系也是一条直角双曲线,那么E-AR关系应该是线性或直角双曲线,推导过程如下:
设、都是关于的直角双曲线函数,即:
其中,、、、均为正数。将从反解出来,得:
将带入到中,得:
针对上式,可能出现3种情况:
(1),此时是一个直角双曲线函数;
(2),此时是一个线性函数;
(3),此时也是一个直角双曲线函数,但是其图像与第(1)种情况关于原点中心对称。
当出现第(3)种情况时,和的均为负值,即和为负值,这显然是不可能的,所以。
当E-AR之间为线性关系时,,此时不存在“受体保留”现象。但实验中发现不同的激动剂在不同的受体占有率下都可以达到,所以这种情况也可以排除。
因此E-AR的关系只能满足直角双曲线这个函数关系,表达式如下:
其中为达到一半所需的的浓度。
2.3.4 传感器函数
E与A的关系也可以写为:
所以为了建立激动剂的量-效关系,我们构建了2个函数关系:(1)受体占据函数,描述从A到AR的过程;(2)传递函数,描述从AR到E的过程。
我们定义传递比率()这一参数:
则:
E-A曲线的渐近线为:
可见,激动剂的最大活性是由,即、这2个参数决定的。
2.4 E-A的非直角双曲线关系
当E-A曲线比直角双曲线更加陡峭或平缓时,E-AR的关系可以用以下表达式:
E-A的关系为:
此时,
渐近线为:
事实上,直角双曲线仅仅是时的特例。